Вступ. Синдром подовженого інтервалу QT (LQTS – Long QT Syndrome), також відомий як синдром Романо-Уорда, є серйозним порушенням серцевого ритму, що виникає внаслідок нерегулярної електричної активності серця. Він визначається подовженим інтервалом QT на електрокардіограмі, що відображає уповільнену реполяризацію клітин серця. Цей стан підвищує ймовірність виникнення небезпечних для життя шлуночкових аритмій, таких як torsades de pointes, які можуть прогресувати до раптової серцевої смерті (РСС) [1, 2]. Шлуночкова недостатність належить до групи каналопатій - захворювань, що виникають через дисфункцію іонних каналів серця. В основі лежить генетична патологія, яка порушує баланс калієвих, натрієвих або кальцієвих струмів через мембрани кардіоміоцитів. Цей дисбаланс впливає на нормальний цикл електричної активності серця, подовжуючи фазу реполяризації і створюючи передумови для тахікардії та фібриляції. Захворювання клінічно і генетично гетерогенне, з численними мутаціями в генах, що кодують білки іонних каналів, що визначає широкий спектр його проявів і складність діагностики [3].
Мета. Вивчити особливості патогенезу, клінічних варіантів, діагностики та лікування хворих на LQTS.
Матеріали і методи. Дослідження включало огляд наукових праць, у тому числі мета-аналізів та клінічних повідомлень з міжнародних кардіологічних журналів, що стосуються LQTS, які були доступні у відкритих наукових базах даних, таких як PubMed, Google Scholar, Кокранівська бібліотека, Національна медична бібліотека та інші.
Результати та обговорення. Етіологія LQTS пов'язана з мутаціями у понад 15 генах, які регулюють функцію іонних каналів у серці. Найчастіше мутації відбуваються в генах KCNQ1, KCNH2 і SCN5A, які відповідають за регуляцію калієвих і натрієвих каналів. Ці генетичні варіації призводять до виникнення декількох підтипів LQTS, серед яких найпоширенішим є LQT1. Викликаний мутаціями в гені KCNQ1, LQT1 становить приблизно 50% всіх діагностованих випадків. У цих пацієнтів напади аритмії зазвичай провокуються фізичним навантаженням, особливо плаванням. LQT2, пов'язана з мутаціями KCNH2, часто провокується емоційним стресом або гучними звуками. LQT3, спричинений мутаціями SCN5A, зазвичай проявляється у стані спокою або під час сну. Мутації інших генів, таких як CALM1, CALM2 або CALM3, пов'язані з рідкісними формами LQTS, які часто супроводжуються більш важкими проявами [2, 4].
Синдром подовженого інтервалу QT можна розділити на генетично гетерогенну вроджену форму та набуту форму, яка часто викликана прийомом ліків. Вроджена форма зустрічається надзвичайно рідко (1:10 000 пологів). Синдром подовженого інтервалу QT має клінічне значення, оскільки як вроджена, так і набута форми синдрому QT можуть призводити до шлуночкової тахікардії[5].
Патогенетично LQTS виникає внаслідок дисфункції іонних каналів, що порушує електролітний баланс у клітинах серця. Порушення функції калієвих каналів затримує відтік іонів калію, що призводить до подовження фази реполяризації. Аналогічно, мутації натрієвих каналів призводять до надмірної активності, що ще більше подовжує процес реполяризації. Характерною ознакою цього розладу є подовжений інтервал QT, що спостерігається на ЕКГ. У деяких випадках у пацієнтів можуть виникати оборотні шлуночкові тахікардії, такі як torsades de pointes, які можуть минати спонтанно або переростати у фібриляцію шлуночків - небезпечний для життя стан, що вимагає негайного медичного втручання [6].
Клінічні прояви LQTS дуже варіабельні. Деякі пацієнти можуть мати безсимптомний перебіг, в той час як у інших можуть спостерігатися непритомність, судоми або навіть РСС. Напади непритомності часто є першими симптомами і виникають через короткочасну гіпоперфузію мозку внаслідок шлуночкової тахікардії. У дітей та підлітків ці симптоми можуть бути помилково діагностовані як епілепсія. Розвиток РСС є найсерйознішим ускладненням, особливо у молодих людей, які не отримували належного лікування. Пусковими факторами таких подій можуть бути фізичні навантаження, емоційний стрес, гучні звуки та певні ліки, такі як макроліди, фторхінолони та антипсихотики [2, 3].
Діагноз LQTS ґрунтується на клінічних даних, ЕКГ та генетичному тестуванні. Основним діагностичним критерієм є подовження QT понад 450 мс у чоловіків і 460 мс у жінок[1].
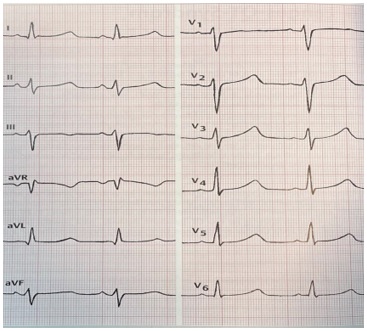
Важливими результатами ЕКГ при синдромі подовженого інтервалу QT є (рисунок):
- Подовження QT або час QT довший за 0,44 секунди (в нормі 0,35-0,44 секунди)
- Шлуночкова тахікардія (torsade de pointes: швидка та поліморфна форма)
- Синусова брадикардія у стані спокою та при фізичному навантаженні
- сплощений або негативний зубець Т
- високий або двофазний зубець U і зубець TU
- залежний від частоти серцевих скорочень час QT
Рис. Синдром подовженого інтервалу QT (синдром Романо-Уорда). ЧСС 90/хв, час QT 0,42 сек, відносний час QT подовжений на 128%, QT патологічно подовжений на 0,49 сек патологічно подовжений[5].
Однак ЕКГ не завжди інформативна, оскільки тривалість інтервалу QT може змінюватися в залежності від частоти серцевих скорочень. Для оцінки ризику використовують спеціальні шкали, такі як шкала Шварца, яка включає показники ЕКГ, симптоми та сімейний анамнез. Генетичне тестування може підтвердити діагноз, визначити підтип LQTS і надати відповідне лікування. Воно також важливе для скринінгу родичів пацієнтів, оскільки синдром передається за аутосомно-домінантним типом [1, 4].
Серед інших причин подовження інтервалу QTc, які слід брати до уваги для проведення диференціальної діагностики, виділяють подовження QT через використання лікарських засобів, гіпокаліємію, певні неврологічні захворювання (зокрема, субарахноїдальну кровотечу), а також структурні захворювання серця. Важливо враховувати ці фактори, щоб уникнути помилкової інтерпретації клінічної картини та забезпечити точну діагностику [7].
Основою лікування LQTS є бета-блокатори, які знижують ризик аритмій, блокуючи адренергічну стимуляцію. Препарати, такі як пропранолол і надолол, є найефективнішими для пацієнтів із LQT1 і LQT2. Пацієнти з LQT3 можуть отримувати додаткову терапію мексилетином, який блокує надмірну активність натрієвих каналів. Для осіб із високим ризиком, наприклад тих, хто переніс зупинку серця, або в кого медикаментозна терапія неефективна, рекомендовано імплантацію кардіовертер-дефібрилятора. У рідкісних випадках може застосовуватись симпатектомія, яка знижує ризик адренергічно залежних аритмій. Пацієнтам також рекомендується уникати провокуючих факторів, зокрема фізичних навантажень, стресу та ліків, що подовжують QT [3, 6].
Висновки. Діагностика пацієнтів з LQTS вимагає надзвичайної пильності та відповідного рівня кваліфікації лікаря через підвищений ризик розвитку РСС, особливо якщо такі пацієнти не отримують належного лікування. Таким чином, високий рівень обізнаності лікарів з проблемою LQTS, своєчасна діагностика та адекватне лікування дозволять попередити розвиток тяжких аритмогенних станів та врятувати життя цієї когорти пацієнтів.
Список використаних джерел
1. Clinical Aspects of Diagnosis and Treatment of Romano-Ward Syndrome [Електронний ресурс]. – Режим доступу: http://mpu.med-expert.com.ua/article/view/250647.
2. Moss A.J., Kass R.S., Schwartz P.J., et al. Long QT Syndrome: Progress in Diagnosis and Therapy // Heart Rhythm. – 2024. – Т. 24, № 12. – Режим доступу: https://www.heartrhythmjournal.com/article/S1547-5271(24)02364-6/fulltext.
3. Piers S.R., Du C.R., Severs N.J., et al. Genetic Basis of Long QT Syndrome: A Review // Journal of Cardiology. – 2022. – Т. 18, № 5. – Режим доступу: https://www.sciencedirect.com/science/article/pii/S2405500X22002985?via%3Dihub
4. Schwartz P.J., Crotti L., Insolia R. Long QT Syndrome: From Genetics to Clinical Management // Heart. – 2022. – Т. 108, № 5. – С. 332–341. – Режим доступу: https://heart.bmj.com/content/108/5/332.long.
5. Cook-Sup So. Praktische EKG-Deutung: Einführung in die Elektrokardiografie. 4th completely revised and expanded edition. – Stuttgart: Thieme Verlag, 2017. – С. 181–183.
6. Milman S., Rudic B., Wright G., et al. Advances in Treatment of Long QT Syndrome // Progress in Cardiovascular Diseases. – 2022. – Т. 70, № 3. – С. 180–188. – Режим доступу: https://www.sciencedirect.com/science/article/abs/pii/S1050173822000901?via%3Dihub.
7. Groffen A. J., Bikker H., Christiaans I. Long QT Syndrome Overview [Електронний ресурс] / A. J. Groffen, H. Bikker, I. Christiaans. – Initial Posting: February 20, 2003; Last Update: March 21, 2024. – Режим доступу: https://www.ncbi.nlm.nih.gov/books/NBK1129/
|