Хвороба Альцгеймера залишається однією з найпоширеніших нейродегенеративних патологій, що суттєво впливає на якість життя пацієнтів. Станом на 2019 рік хворих із втратами когнітивних здібностей нараховувалося близько 55 мільйонів випадків у всьому світі, переважно це люди похилого віку. Прогнози вказують на те, що до 2050 року ця цифра зросте майже втричі [1].
Розробка нових терапевтичних засобів, спрямованих на інгібування ключових молекулярних мішеней є пріоритетом сучасної фармакології. Методи in silico мають суттєві переваги над методами in vivo та in vitro на ранніх етапах пошуку нових терапевтичних сполук завдяки їхній ефективності, економічності та високій продуктивності. Зокрема, in silico підходи, як-от віртуальний скринінг і молекулярний докінг, дозволяють швидко аналізувати великі бази даних хімічних сполук, оцінювати їх потенційну взаємодію з біологічними мішенями та прогнозувати ключові фармакологічні властивості. Це значно скорочує витрати часу та коштів порівняно з фізичними експериментами.
Метою роботи є виявлення потенційних низькомолекулярних сполук для терапії хвороби Альцгеймера шляхом проведення комп'ютерного скринінгу та оцінка їхньої здатності взаємодіяти з ключовими біологічними мішенями методом молекулярного докінгу.
До відомих макромолекулярних мішеней, залучених у патологію хвороби Альцгеймера відносяться: ацетилхолінестераза, бета-секретаза, гама-секретаза, каспази, ацетилхолінові рецептори, NMDA-рецептори, ферменти ROCK-I та
NOX2 [2].
Розробка інгібіторів β-секретази стикається з труднощами через складність подолання кандидатами гематоенцефалічного бар'єру, побічні ефекти, пов’язані з фізіологічними функціями β-секретази та відсутність доказів значного клінічного ефекту у зменшенні прогресування захворювання. У той же час інгібітори AChE вже демонструють клінічно значущі результати у покращенні когнітивних функцій, оскільки їхній механізм дії спрямований на симптоматичне лікування [3]. П’ять із семи схвалених FDA препаратів для терапії хвороби Альцгеймера є інгібіторами ацетилхолінестерази.
При виборі комплексу білка-мішені з інгібітором для проведення комп'ютерного скринінгу в контексті розробки лікарських препаратів враховують низку критеріїв, таких як роздільна здатність комплексу, достовірність координат ліганду, значущість білка-мішені, доведена біологічна активність ліганду та інші. Одним із найважливіших є доступність структурної інформації в хорошій роздільній здатності (<3,0 Å) [4]. Найбільшою бібліотекою 3D-структур білків є Protein Data Bank (PDB), зокрема там міститься структурна інформація про 71 комплекс із ацетилхолінестеразою людини з різними лігандами.
Для пошуку потенційних терапевтичних сполук було використано методи віртуального скринінгу на основі фармакофора. Для побудови моделі фармакофора та проведення скринінгу обрано сервіс Pharmit.
Для докінгу використано сервіс SwissDock. SwissDock використовує два основні алгоритми для докінгу: Attractive Cavities і AutoDock Vina.
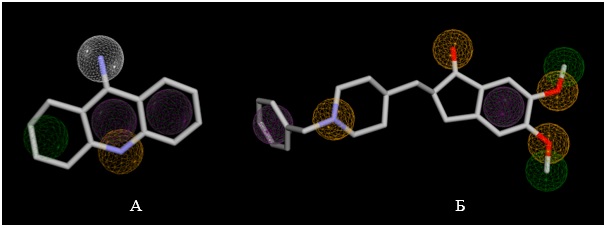
Для дослідження було обрано білково-лігандні комплекси ацетилхолінестерази з такрином (PDB ID: 7XN1; комплекс 1) та з донепезилом (PDB DI: 7E3H; комплекс 2). На основі цих комплексів було створено фармакофорні моделі (рисунок 1), що враховують ключові взаємодії білка з лігандом. Модель для комплексу 1 містить 7 ознак (1 донор водневого зв’язку, 3 гідрофобні групи, 2 ароматичні групи та 1 акцептор водневого зв’язку), для комплексу 2 – 10 ознак (2 ароматичні групи, 4 акцептори водневого зв’язку, 4 гідрофобні групи.
А – Фармакофорна модель для такрину; Б – фармакофорна модель для донепезилу.
Рисунок 1 – Фармакофорні моделі інгібіторів, побудовані з використанням сервісу Pharmit.
Результати віртуального скринінгу було відфільтровано з використанням правил лікоподібності (п’яти Ліпінського). Також відбиралися сполуки, для ких середньоквадратичне відхилення позицій атомів (RMSD) становило менше 1,0 Å, а оціночна функція в сервісі було від’ємною. RMSD дозволяє оцінити геометричну схожість сполук з фармакофором, оціночна функція може дати приблизну інформацію про прогнозовану силу зв’язування.
У результаті віртуального скринінгу із застосуванням цих моделей відібрано 548 сполук для комплексу 1 та 186 сполук для комплексу 2.
Для подальшої оцінки для 20 найкращих сполук, обраних за значеннями RMSD та оціночної функції, було проведено молекулярний докінг.
Молекулярний докінг обраних сполук виявив найкращі значення енергії зв’язування для PubChem-60440705 (афінність зв’язування за AutoDock Vina -9,521 ккал/моль, оціночна фкнкція Attractive Cavities -2,122407) і PubChem-63351446 (афінність зв’язування за AutoDock Vina -6,885 ккал/моль, оціночна функція Attractive Cavities -21,454583). Відсутність досліджень про їхню інгібуючу активність у літературі свідчить про новизну результатів, що робить ці сполуки перспективними кандидатами для подальших експериментальних досліджень, таких як оцінка їхньої інгібуючої активності in vitro та in vivo.
Отримані результати дозволяють розширити перелік перспективних кандидатів для терапії хвороби Альцгеймера. Ідентифіковані сполуки можуть бути використані для подальшого експериментального підтвердження їхньої біологічної активності, що створює передумови для розробки нових лікарських засобів.
1. Alzheimer’s Disease International, World Alzheimer Report 2019: Attitudes to dementia. 2019.
2. Advances in Applying Computer-Aided Drug Design for Neurodegenerative Diseases / M. M. Salman et al. International Journal of Molecular Sciences. 2021. Т. 22, № 9. С. 4688.
3. Efficacy of acetylcholinesterase inhibitors in Alzheimer's disease / G. Marucci et al. Neuropharmacology. 2020. С. 108352.
4. Моделювання молекулярної взаємодії [Електронний ресурс] : підручник для здобувачів ступеня магістра за спеціальністю 162 «Біотехнології та біоінженерія» / С. В. Кисляк, Голуб Н. Б., Дуган О. М., Аверьянова О. А. ; КПІ ім. Ігоря Сікорського. – Електронні текстові дані (1 файл, 26 Мбайт). – Київ : КПІ ім. Ігоря Сікорського, 2023. – 203 с.
|